Projects
Selected projects in Bioinformatics and Computational Biology, with a focus on data analysis, tool development, and applied solutions.
Machine learning in microbial genomics
Machine learning and pipeline development for large-scale microbial genome analysis
Research project in the Microverse Cluster at the University of Jena, within the Viral Ecology and Omics group, led by Prof. Dr. Bas E. Dutilh.
I analyzed 91,228 microbial genomes to study adaptation to environmental conditions. For this, I developed 24 optimal classification and regression Machine Learning models (linear regression, Random Forest, k-nearest neighbours, naive Bayes, Support Vector Machines). They can predict salinity, temperature, oxygen and pH levels for bacterial growth in the lab and can assist experimental scientists cultivating new isolates. I found specific protein coding genes and ncRNAs that could potentially be used for biotechnological applications. I also developed FxTractor, a scalable pipeline for high-throughput genomic feature extraction.
My roles in this project were to lead the data analysis, develop models and tools and coordinate collaborations. Relevant techniques I used were: Machine Learning, Bioinformatics, python, snakemake, HPC (slurm), R, bash, Linux, conda, Git.
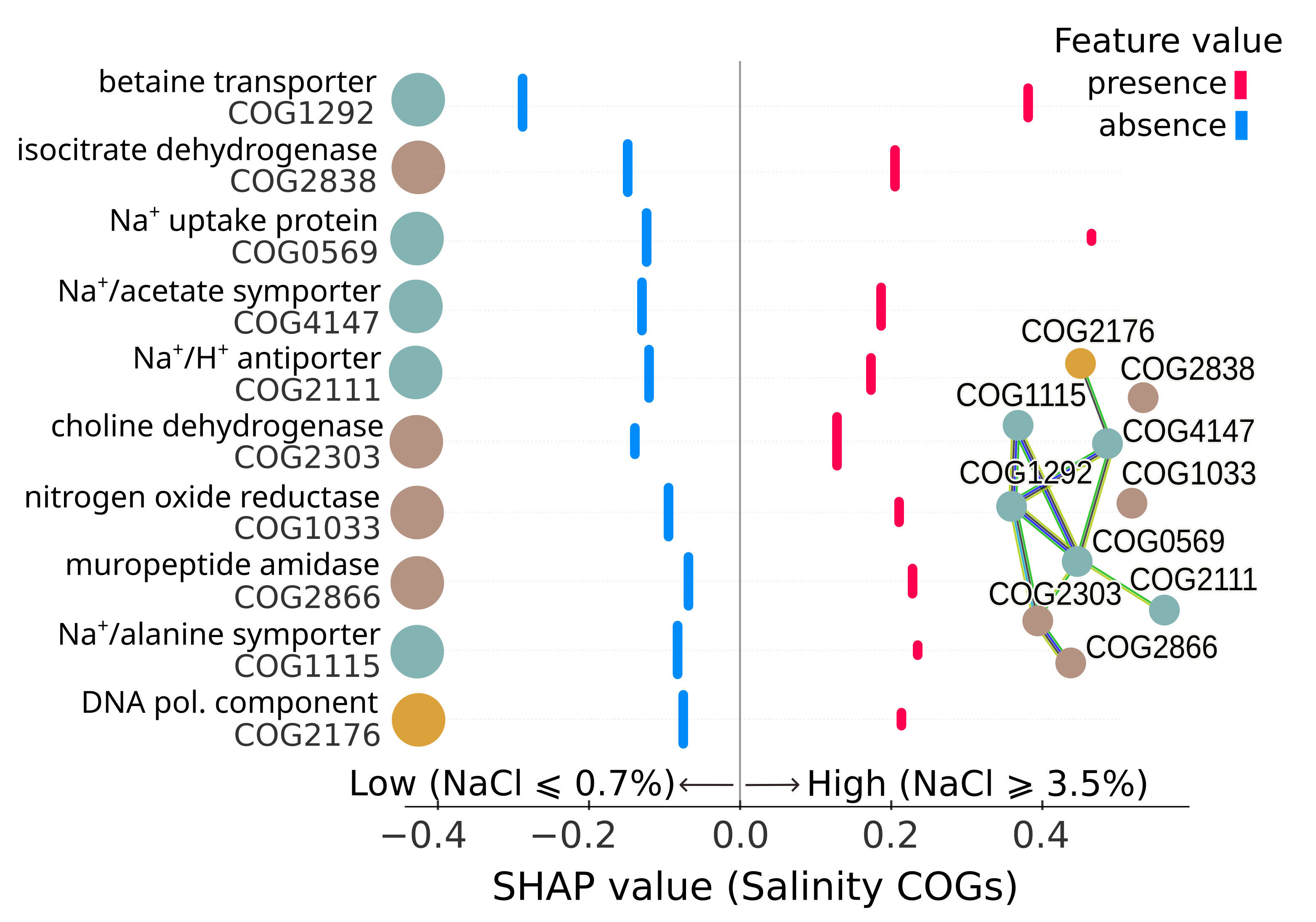
Figure: Top 10 most important proteins (COGs) in ML classification models of microbes with salinity preferences above the ocean's average salinity (~3.5% NaCl). Plot shows Shapley additive explanations (SHAP) scores. Network shows functional connections.
Results
• 1 published scientific article (Structural color in bacteria) and 2 in development
• 2 Bioinformatics tools released on GitHub (FxTractor and
Visualization of gene synteny)
• Co-supervision of a PhD student
• Co-design of a Masters-level course: Big data analysis and interpretation
Clinical data analysis
Statistical analysis of laboratory data and decision support for increasing patient safety
Clinical data analysis at the University of Leipzig Medical Center, within the AMPEL work group, led by Prof. Dr. Med. Torsten Kaiser. This work was done in collaboration with clinicians and industry partners from Xantas AG and had as aim the development of the Clinical decision Support System (CDSS) AMPEL.
I developed three computational projects using statistical methods in R on SAP-based clinical data. The aim was at fast diagnostics of anemia, hypofibrinogenemia and end-stage liver disease (MELD). I also structured and standardized the knowledge dataflow of the University of Leipzig Medical Center, enabling consistent downstream analysis and integration into decision support systems. My work contributed to the development of the AMPEL CDSS system, which is designed to assist physicians in taking quick actions with fast diagnosis.
My roles in these projects were to perform data analysis, collaborate closely with physicians and industry partners, supporting decision-making. Relevant techniques I used were: Statistics, R, bash, Linux, Git.
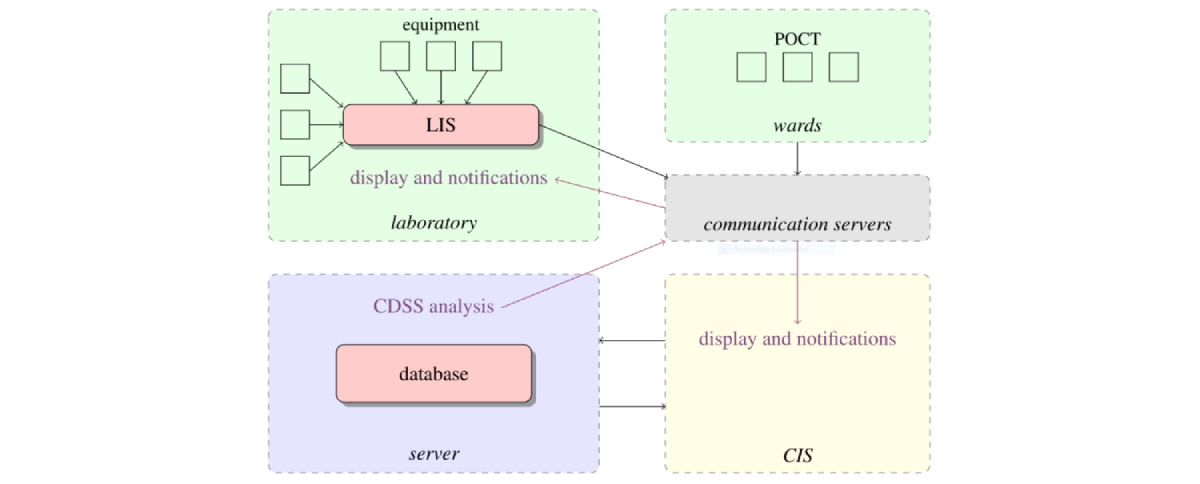
Figure: Conceptual representation of the knowledge dataflow of the University of Leipzig Medical Center including the CDSS system AMPEL, which performs fast clinical diagnosis. When a diagnosis, such as acute anemia, is detected in the wards, AMPEL identifies it and sends messages to nurses and clinicians for fast action to improve patient safety.
Results
• 4 published scientific articles (AMPEL Framework,
MELD,
Anemia,
Metabolic Syndrome)
• Data-driven contributions to the AMPEL CDSS system used in hospital diagnostics
• Improved consistency and usability of clinical laboratory data
• Close collaboration with clinicians and cross-functional teams
Computational analysis of ncRNAs
Methods and tool development for evolutionary analysis of ncRNAs
Doctoral project at the University of Leipzig, Faculty of Mathematics and Computer Science, Chair of Bioinformatics, supervised by Prof. Dr. Peter Stadler and Prof. Dr. Katja Nowick.
I developed novel algorithms and statistical methods to detect adaptive selection in non-coding RNA (ncRNA) structures. I designed and implemented the SSS-test, the first method to identify positive selection in RNA secondary structures by integrating sequence and structural information (Perl, Bash, R). I applied the test to primate datasets to identify candidate human lncRNAs under adaptive evolution, enabling scalable analysis of RNA function with potential applications in genomics.
My roles in this project were to lead the data analysis, develop methods and tools and coordinate collaborations. Relevant techniques I used were: Bioinformatics, Statistics, perl, bash, Linux, Git, HPC (SGE).
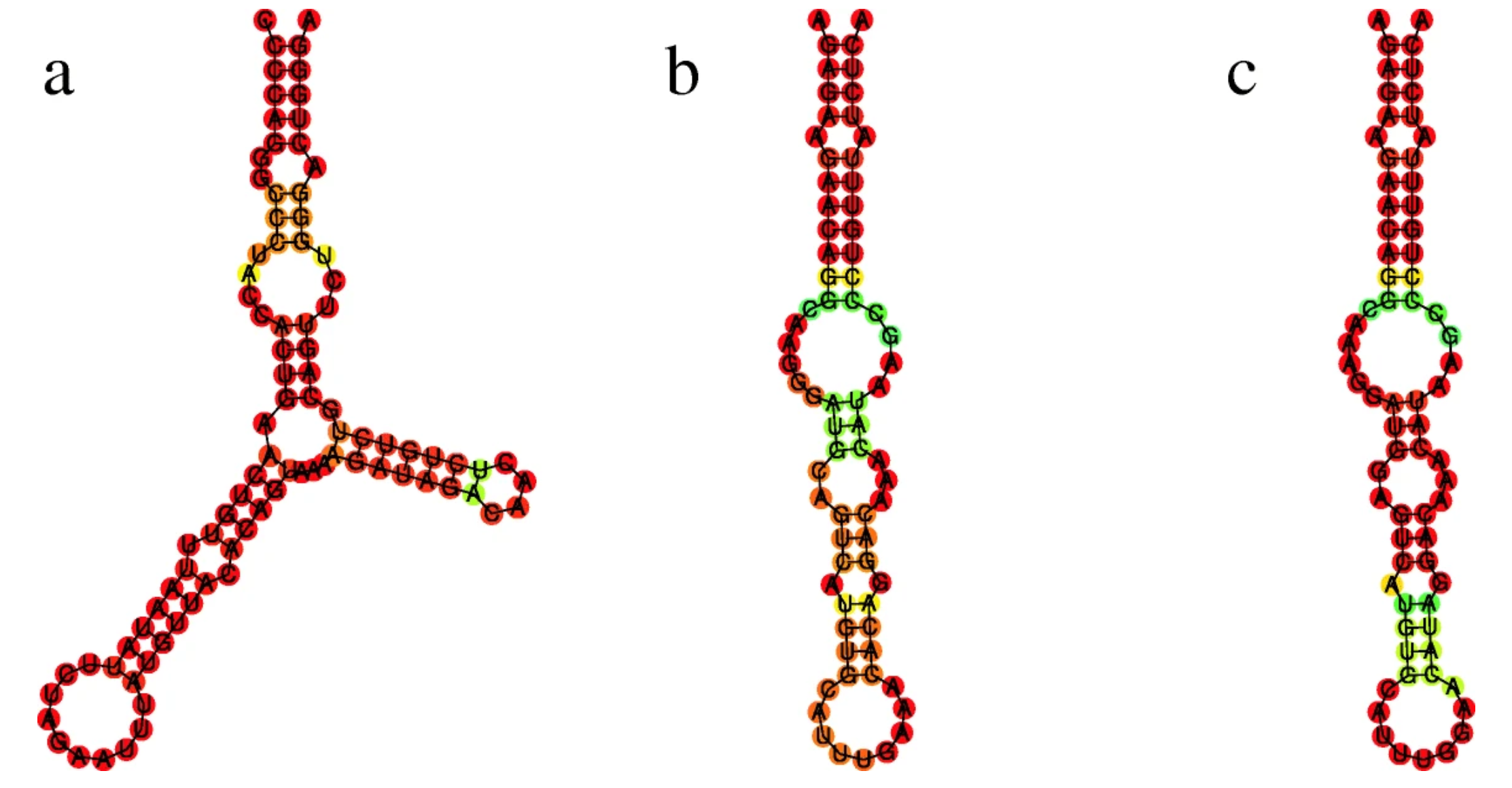
Figure: Example result from the SSS-test. The lncRNA LINC02217 shows evidence of positive selection in humans (a) compared to (b) chimpanzees and (c) gorillas. I identified this structure as one of 111 candidates under adaptive selection from a set of 87,613 analyzed structures across primate genomes.
Results
• 6 published scientific articles (SSS-test,
RNA evolution,
Visualization of RNAs,
Genomic evolution of reptiles,
HAR1 ncRNA evolution,
Genomic evolution in humans)
• 1 Book chapter (Evolutionary conservation of RNAs)
• 2 Bioinformatics tools released on GitHub (SSS-test and
Long ncRNA orthology)
• Doctoral dissertation in Bioinformatics: "Adaptive Evolution of Long Non-Coding RNAs"
• Coordination of multidisciplinary and multicultural collaborations
Immunogenomics analysis
Statistical and NGS analysis of antibody–DNA interactions
Master’s project at the University of Brasília (Brazil), Institute of Biological Sciences , Graduation Program of Molecular Biology, supervised by Full Prof. Dr. Marcelo Brígido.
I analyzed high-throughput Illumina sequencing data of antibody repertoires (VH4 and VH10 families) to investigate DNA-binding properties. Using classical and Bayesian statistical methods, I found that heavy chains of antibodies confer antigen recognition properties. My results showed that VH10 antibodies have intrinsic DNA-binding capacity, providing novel insight into antibody recognition mechanisms. This could open new doors for research in systemic lupus erythematosus, a medical condition that is characterized by the presence of anti-DNA antibodies.
My roles in this project were to lead the data analysis and interpret results in a biological context. Relevant techniques I used were: Statistics (classical and Bayesian), Bioinformatics, perl, bash, NGS sequence analysis.
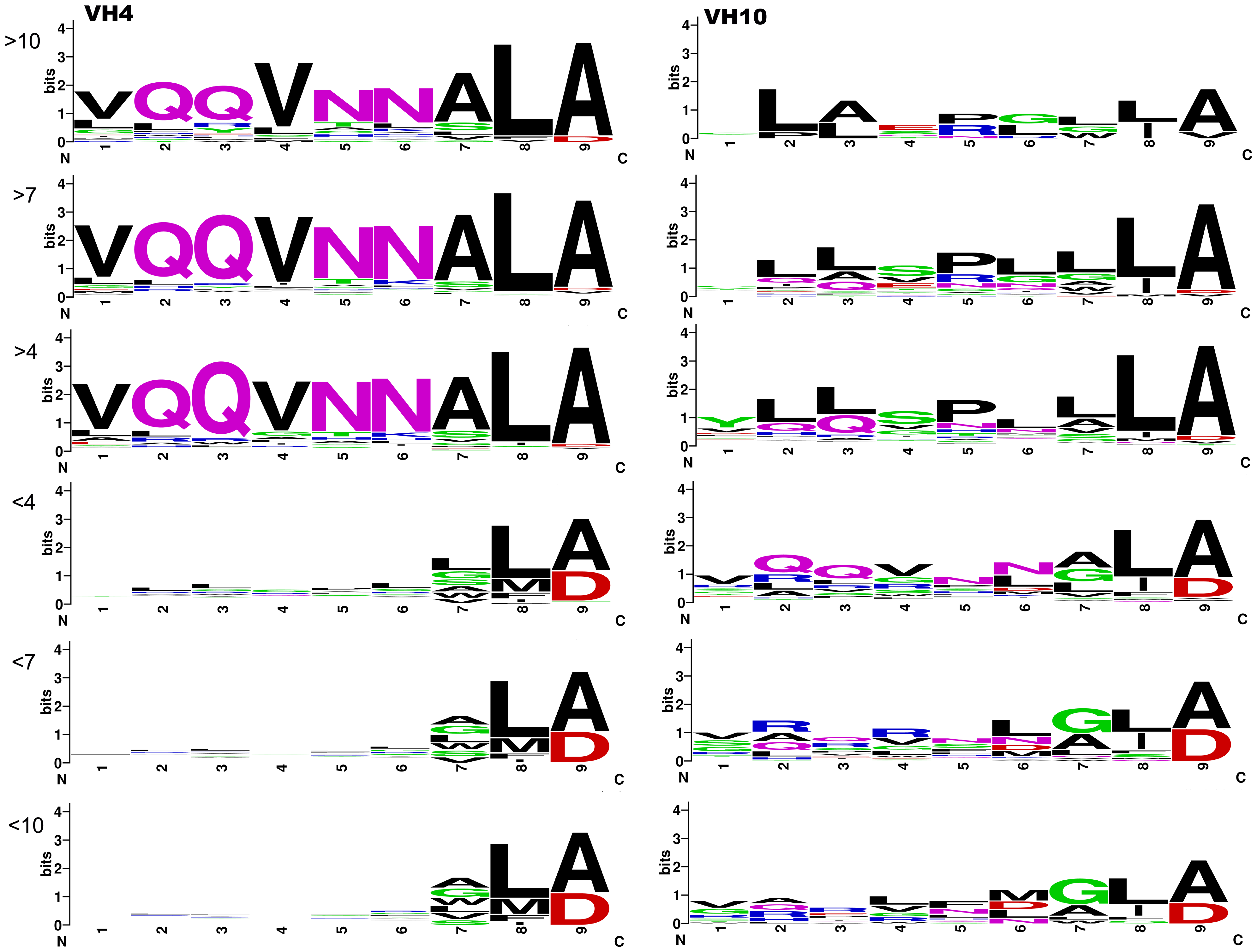
Figure: VH10 antibodies (right) show higher diversity in the CDR3 region and intrinsic DNA-binding capacity, while VH4 antibodies (left) exhibit sequence-specific binding patterns.
Results
• 1 published scientific article (Anti-DNA antibodies)
• Masters thesis in Molecular Biology: "Genetic Variability of anti-DNA antibodies"
• Provided insights into antibody repertoire formation and immune response